

Plicní arteriální hypertenze - PAH Plicní venookluzivní nemoc a plicní kapilární hemangiomatóza Perzistující plicní hypertenze novorozenců - PPHN Plicní hypertenze při postižení levého srdce Plicní hypertenze při plicních onemocněních a/nebo při hypoxémii Chronická tromboembolická plicní hypertenze
Chronická plicní hypertenze
Plicní hypertenze je syndrom hemodynamicky charakterizovaný zvýšením středního tlaku v plicnici ≥ 25 mmHg. Plicní cévní rezistence není součástí obecné definice syndromu plicní hypertenze. Naopak je součástí hemodynamické definice plicní arteriální hypertenze (PAH), která zahrnuje střední tlak v plicnici ≥ 25 mmHg, tlak v zaklínění ≤ 15 mmHg, plicní cévní rezistenci ≥ 3 WU (Woodovy jednotky) a současně normální nebo nižší srdeční výdej. U pacientů s vyšším srdečním výdejem (levo-pravé zkraty, jaterní cirhóza) je již hodnota plicní cévní rezistence nad 1.5 WU abnormální, ale automaticky nemusí svědčit pro PAH. Hodnoty středního tlaku v plicnici 21-24 mmHg představují hraniční tlaky v plicnici. Zejména u pacientů se systémovou sklerodermií a s rodinnou anamnézou idiopatické nebo hereditární PAH jejich nález představuje významný rizikový faktor progrese do plicní hypertenze.
Z dostupných dat stále nemáme k dispozici dostatečnou evidenci pro definici zátěžových kritérií plicní hypertenze.
Plicní hypertenze vzniká jako důsledek mnoha onemocnění plic, srdce nebo v souvislosti s poruchami regulace dýchání. Patofyziologická klasifikace rozděluje plicní hypertenzi podle mechanismu vzniku na prekapilární (zvýšený tlak v plicnici, normální v zaklínění) a postkapilární (zvýšený tlak v plicnici i v zaklínění), a tu dále na izolovanou a kombinovanou. Prekapilární plicní hypertenze je hlavním důvodem vzniku izolované hypertrofie pravé komory srdeční, tzv. chronického cor pulmonale. Klinická klasifikace rozeznává sedm kategorií plicní hypertenze. Klinické jednotky v každé kategorii mají do jisté míry podobnou patogenezi, histologický obraz, kliniku a léčbu.
Plicní arteriální hypertenze – PAH (skupina 1)
Definice
PAH je chronické, progresivní a potenciálně fatální onemocnění plicního oběhu vedoucí k selhání pravé komory srdeční. Z hlediska hemodynamické definice jde o prekapilární plicní hypertenzi se zvýšenou plicní cévní rezistenci ≥ 3 WU a současně s normálním nebo nižším srdečním výdejem.
Do skupiny PAH je řazena především idiopatická PAH, u níž dosud neznáme vyvolávající faktor. U hereditární formy není vyvolávající faktor rovněž znám. Onemocnění se však dává do souvislosti s některými mutacemi. Dále patří do skupiny PAH řada stavů, kde je vyvolávajícím faktorem systémové onemocnění pojiva, vrozená zkratová srdeční vada, portální hypertenze, abúzus anorektik nebo HIV infekce. Ze skupiny PAH jsou vyčleněny stavy spojené s postižením venul nebo plicních kapilár (plicní venookluzivní nemoc, plicní kapilární hem¬angiomatóza) a dále perzistující plicní hypertenze novorozenců.
Idiopatická a hereditární PAH představuje zhruba 43 % populace PAH. Její minimální prevalence v dospělé evropské populaci se odhaduje nejméně na 15 případů na milion dospělých obyvatel. Idiopatická PAH se v populaci vyskytuje s roční incidencí 2-5 případů na milion obyvatel, častěji jsou postiženy ženy. Hereditární forma tvoří 6-10 % případů PAH. Asi v 80 % těchto nemocných je přítomna mutace v genu BMPR2 (bone morphogenetic protein receptor 2), který patří do skupiny receptorů pro transformující růstový faktor beta. Jedná se pak o onemocnění přenášené autozomálně dominantně s variabilní penetrancí (jen 10-20% nositelů mutace má projevy onemocnění) a expresivitou. Zdá se, že je přítomna tzv. genetická anticipace, kdy u každé následující postižené generace nastává postižení dříve a/nebo je klinicky závažnější. U idiopatické PAH může být přítomna mutace BMPR2 přibližně u 10-20% pacientů a v tomto případě se pravděpodobně jedná o nové mutace, které mohou být ale dále předány potomkům. Asi u 5 % pacientů s hereditární PAH je přítomna mutace v dalších genech patřících do skupiny receptorů pro transformující růstový faktor beta: ALK 1, ENG, SMAD 9, CAV 1 , KCNK 3. Asi ve 20 % rodin s hereditární PAH není detekovatelná mutace v dosud známých genech spojených s rozvojem onemocnění.
PAH při abúzu některých léků a toxických látek (některá anorektika, amfetamin, metamfetamin, kokain) pravděpodobně souvisí s interakcí s nitrobuněčným transportem serotoninu, případně dopaminu a norepinefrinu. Elevace tlaků v plicnici může být patrna již po 3-4 týdnech užívání anorektik, k rozvoji PAH je však třeba zpravidla více jak šesti měsíců. K nově identifikovaným rizikovým faktorům rozvoje PAH se počítá anorektikum a hypolipidemikum benfluorex, desatinib užívaný v léčbě chronické myeloidní leukémie v případě rezistence na imatinib a interferon alfa a beta. Užívání inhibitorů zpětného vychytávání serotoninu těhotnými v pokročilém stádiu gravidity zvyšuje riziko perzistující plicní hypertenze novorozenců.
PAH u nemocných se systémovými onemocněními pojiva představuje přibližně 15-20 % případů PAH v dospělé populaci. Systémová sklerodermie, zejména její CREST varianta, je nejčastější příčinou PAH mezi systémovými onemocněními. Prevalence PAH u nemocných se systémovou sklerodermií kolísá nejčastěji mezi 7 a 12 %. U systémového lupus erytematodes je plicní hypertenze přítomna u 5-10 % pacientů. Vzácněji se PAH vyskytuje u revmatoidní artritidy, dermatomyositidy, polymyositidy a Sjögrenova syndromu. Histologický obraz u PAH při systémových onemocněních pojiva se zásadně neliší od obrazu u idiopatické PAH.
PAH asociovaná s HIV infekcí se vyskytuje asi u 0,5 % infikovaných nemocných. V patofyziologii se předpokládá působení cytokinů, růstových faktorů a endothelinu. V aktivaci zánětlivých mechanismů hrají zřejmě zásadní roli některé proteiny viru HIV (proteiny Tat, Nef).
U PAH při portální hypertenzi může být jedním z klíčových mechanismů přestup vazoaktivních mediátorů (např. serotonin produkovaný enterochromafinními buňkami ve střevě) z portální do plicní cirkulace při otevřených porto-systémových zkratech. Až 20 % pacientů před transplantací jater může mít střední tlak v plicnici vyšší než 25 mmHg, především jako důsledek vyššího srdečního výdeje. Plicní cévní rezistence bývá zvýšena jen u menšiny z nich. PAH se vyskytuje u 1-6 % nemocných s portální hypertenzí. Riziko vzniku PAH roste s délkou trvání portální hypertenze. Hodnoty středního tlaku v plicnici nad 35 mmHg a současně zvýšená plicní cévní rezistence nad 3 WU představují relativní kontraindikaci transplantace jater. Naopak i středně těžká až těžká plicní hypertenze nemusí být kontraindikací transplantace jater, pokud je na podkladě zvýšeného srdečního výdeje.
PAH asociovaná s vrozenými srdečními vadami souvisí nepochybně významně s recirkulací, přesto představuje jen část problematiky plicní hypertenze u vrozených srdečních vad, které se mohou komplikovat vedle PAH i jinými typy plicní hypertenze. PAH u vrozených srdečních vad zahrnuje:
- Eisenmengerův syndrom, který představuje extrémní formu plicní cévní choroby s těžkou ireverzibilní PAH, která vzniká u některých zkratových vad, není-li velký zkrat uzavřen do 1 roku věku. U Eisenmengerova syndromu bývá tlak v plicnici vyšší než 90% systémového tlaku, plicní cévní rezistence je vysoká (obvykle > 7 WU) a plicní průtok není významně zvýšen oproti systémovému. Zkrat je bidirekční, u otevřené tepenné dučeje může být pouze pravo-levý při suprasystémovém tlaku v plicnici. Prognóza pacientů s Eisenmengerovým syndromem je horší než u běžné populace i než u jiných vrozených srdečních vad, avšak je významně lepší než u idiopatické PAH. Padesáti let věku se dožívá 55 % pacienů s Eisenmengerovým syndromem.
- Hyperkinetická plicní hypertenze se zvýšeným tlakem v plicnici, který odpovídá vysokému průtoku plicním řečištěm u levo-pravých zkratů, plicní cévní rezistence je normální nebo jen mírně zvýšená. U starších pacientů s významným levo-pravým zkratem bývá již plicní cévní rezistence mírně zvýšená (obvykle do 4 – 5 WU) a plicní hypertenze může být těžká. Pokud však převažuje levo-pravý zkrat s vysokým plicním průtokem (Qp/Qs>1,5), nejedná se o Eisenmengerův syndrom. Typicky se vyskytuje u defektů síňového septa typu II, u většiny inkompletních defektů atrioventrikulárního septa a u některých defektů komorového septa nebo otevřených tepenných dučejí.
- Pozdní pooperační PAH vzniká tehdy, je-li zkratová vada uzavřena příliš pozdě, v době, kdy již došlo k remodelaci plicního cévního řečiště. Plicní hypertenze přetrvávat i po uzávěru zkratu, vzácně může i progredovat. Hemodynamika se podobá idiopatické PAH, pacienti nemají cyanózu. Nejčastěji se vyskytuje u pozdního uzávěru defektu komorového septa nebo u komplexních vrozených srdečních vad.
- Těžká PAH u malých defektů se vyskytuje vzácně u pacientů s hemodynamicky nevýznamnou zkratovou vadou, např. u malých defektů síňového septa typu II. Může se jednat o koincidenci s PAH.
PAH asociovaná se schistosomiasou představuje z hlediska celosvětového výskytu zřejmě nejčastější příčinu PAH. Vyskytuje se u 5 % nemocných se schistosomiasou, která postihuje játra a slezinu (10 % z asi 200 milionů osob postižených schistosomiasou). Hemodynamika je obdobná jako u PAH asociované s portální hypertenzí.
Medián přežití u neléčené idiopatické PAH je 2,8 roku. Medián přežití u neléčené PAH při systémové sklerodermii se pohybuje kolem 12 měsíců. Podobně nepříznivou prognózu má PAH asociovaná s HIV infekcí. Naopak lepší prognózu než u idiopatické PAH pozorujeme u nemocných s PAH asociovanou s vrozenou srdeční vadou.
Příznaky u nemocných s plicní hypertenzí nejsou specifické a často se vyskytují až při zvýšení tlaku v plicnici na dvojnásobek normálních hodnot a především při poklesu srdečního výdeje. Právě nespecifické projevy onemocnění jsou příčinou tak časté pozdní diagnózy.
Nejčastějším symptomem je postupně progredující námahová dušnost a únavnost. Závažnost dušnosti významně koreluje s prognózou. Anginózní bolesti na hrudi jsou důsledkem ischémie pravé komory, synkopy a presynkopy projevem nízkého srdečního výdeje. Mezi vzácnější projevy onemocnění patří chrapot způsobený útlakem levého vratného nervu dilatovaným kmenem plicnice, kašel a hemoptýza.
Ve fyzikálním nálezu souvisí manifestace jednotlivých nálezů s tíží plicní hypertenze. Často bývá akcentace druhé srdeční ozvy nad plicnicí, přítomnost čtvrté ozvy a cvalového rytmu. Třetí ozva bývá přítomna v pokročilých stádiích onemocnění. Může být slyšitelný šelest trikuspidální a pulmonální regurgitace. U většiny nemocných je zvýšená náplň krčních žil a hmatná systolická pulzace v prekordiu a v epigastriu při hypertrofii pravé komory. Známkou pokročilého onemocnění je přítomnost periferních otoků, ascitu a cyanózy.
Diagnostika
Cílem jednotlivých vyšetřovacích metod u nemocného s podezřením na plicní hypertenzi je plicní hypertenzi potvrdit nebo vyloučit, v případě průkazu ji kvantifikovat a zjistit její původ.
Diferenciální diagnostika PAH spočívá ve vyloučení častějších příčin syndromu plicní hypertenze (obr. 1).

Obr. 1: Diagnostický algoritmus chronické plicní hypertenze
Echokardiografie je klíčovým vyšetřením v detekci plicní hypertenze (obr. 2,3).
Pro odhad stupně plicní hypertenze je nezbytné dopplerovské echokardiografické vyšetření.
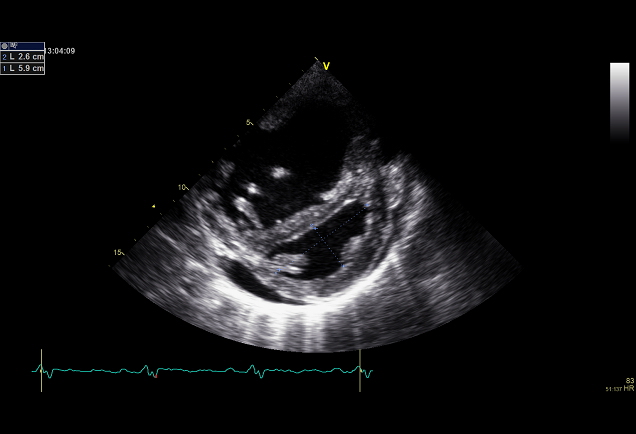
Obr. 2: Echokardiografický obraz perikardiálního výpotku za spodní stěnou levé komory a D tvar levé komory v parasternální projekci na krátkou osu u nemocného s těžkou PAH
Laskavě zapůjčil Prof. MUDr. Aleš Linhart, DrSc., II. interní klinika kardiologie a angiologie VFN a 1.LF UK, Praha
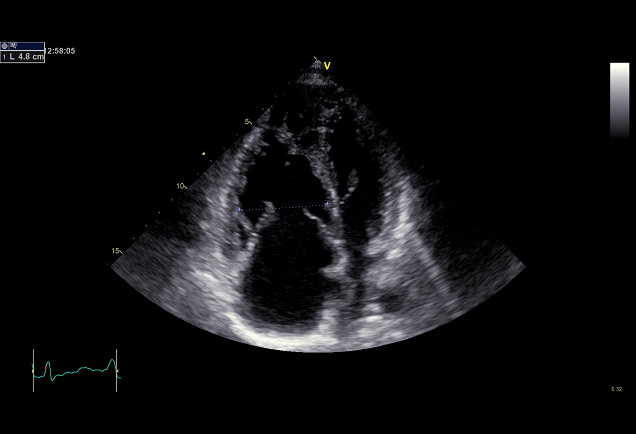
Obr. 3: Laskavě zapůjčil Prof. MUDr. Aleš Linhart, DrSc., II. interní klinika kardiologie a angiologie VFN a 1.LF UK, Praha
Přítomnost plicní hypertenze je nepravděpodobná, pokud odhadovaný systolický tlak v plicnici je menší nebo roven 36 mmHg a současně nejsou přítomny jiné echokardiografické známky plicní hypertenze jako zkrácení akceleračního času na plicnici, dilatace pravostranných srdečních oddílů, hypertrofie volné stěny pravé komory, D tvar levé komory.
Přítomnost plicní hypertenze je možná, pokud je odhadovaný systolický tlak v plicnici 37–50 mmHg nebo pokud je systolický tlak v plicnici ≤ 36 mmHg a současně jsou přítomny jiné známky svědčící pro plicní hypertenzi.
Plicní hypertenze je pravděpodobná, pokud je systolický tlak v plicnici ≥ 50 mmHg.
Je-li na základě echokardiografického vyšetření přítomnost plicní hypertenze možná nebo pravděpodobná, a současně je přítomno postižení myokardu nebo chlopní levého srdce, jedná se pravděpodobně o plicní hypertenzi při onemocnění levého srdce. Je-li oblast levého srdce normální, je nutno vyloučit zkratovou srdeční vadu (echokardiografické vyšetření s kontrastem, případně jícnová echokardiografie). Svědčí-li nález pro zkratovou srdeční vadu, může se jednat plicní arteriální hypertenzi asociovanou s vrozenou zkratovou srdeční vadou a podezření je třeba ověřit katetrizačně.
Je-li nález kontrastní nebo jícnové echokardiografie bez podezření na zkratovou vadu, je nezbytné vyšetření plicních funkcí. V případě jejich výrazné redukce (FVC nebo TLC nebo FEV1 < 60 % náležitých hodnot) je nutno pomocí HRCT a polysomnografie vyloučit plicní hypertenzi řazenou do skupiny 3 klinické klasifikace. U nemocných s PAH může být redukována vitální kapacita zhruba na 80 % náležitých hodnot a difúzní kapacita pro CO asi na 40-80 % náležitých hodnot. Izolovaná redukce difúzní kapacity pro CO bez proporcionální redukce plicních objemů je nálezem značně charakteristickým pro přítomnost plicní hypertenze.
V případě nevýznamné redukce ventilačních parametrů představuje další krok provedení ventilační a perfúzní scintigrafie plic. Je- li nález normální nebo jsou-li přítomny nesegmentární defekty perfúze, je indikována pravostranná srdeční katetrizace k průkazu možné PAH. Součástí katetrizačního vyšetření je pak také provedení testu akutní plicní vazodilatace (adenozinem, epoprostenolem nebo NO).
V případě segmentárních perfúzních defektů na plicním scanu je indikována CT angiografie, angiografie a pravostranná srdeční katetrizace k průkazu možné CTEPH.
Na EKG mohou být známky hypertrofie pravé komory. Jedná se však o nález pro plicní hypertenzi specifický, ale málo senzitivní. Dále může být na EKG přítomna blokáda pravého raménka Tawarova, denivelace úseku ST, abnormity vlny T a P (obr. 4).
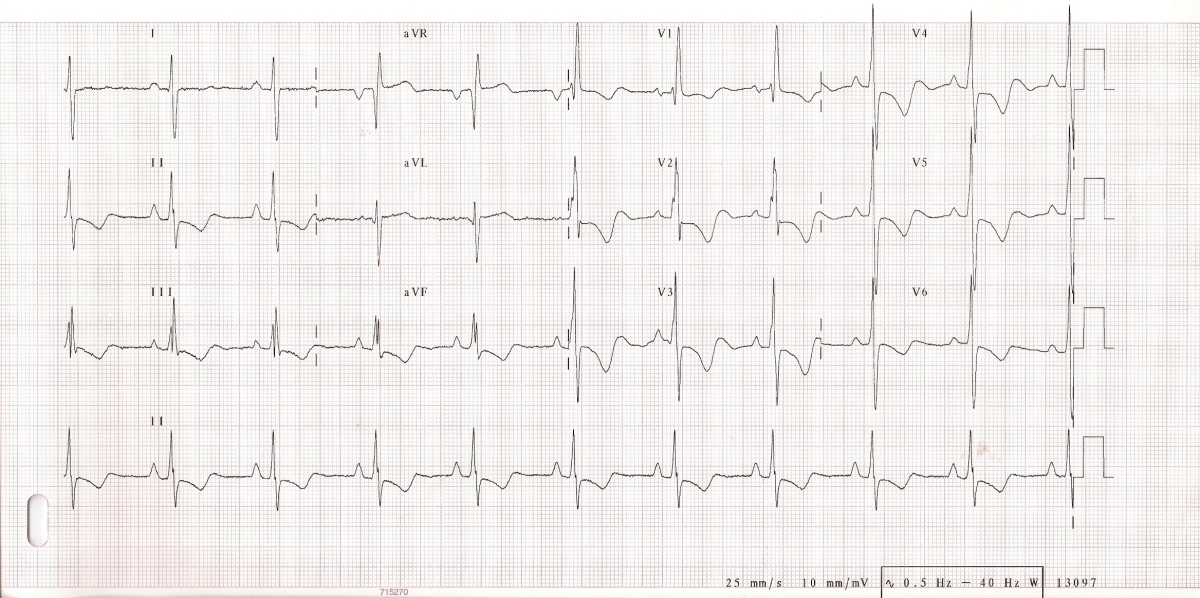
Obr. 4: EKG u nemocné s těžkou plicní arteriální hypertenzí: P pulmonale. R/S ve V1>1, inkompletní blokáda pravého raménka Tawarova, naznačené deprese ST a negativní T ve svodech II, III, aVF, V1-6
Na RTG snímku hrudníku bývá u PAH přítomna dilatace kmenů plicnice a náhlé zúžení cév na hranici lalokových a segmentárních tepen. Důsledkem spasmu periferních plicních cév je zvýšení transparence periferie plicních polí. U pokročilého onemocnění dochází k rotaci srdce proti směru hodinových ručiček (obr. 5). Nález na snímku hrudníku však může být také zcela normální.
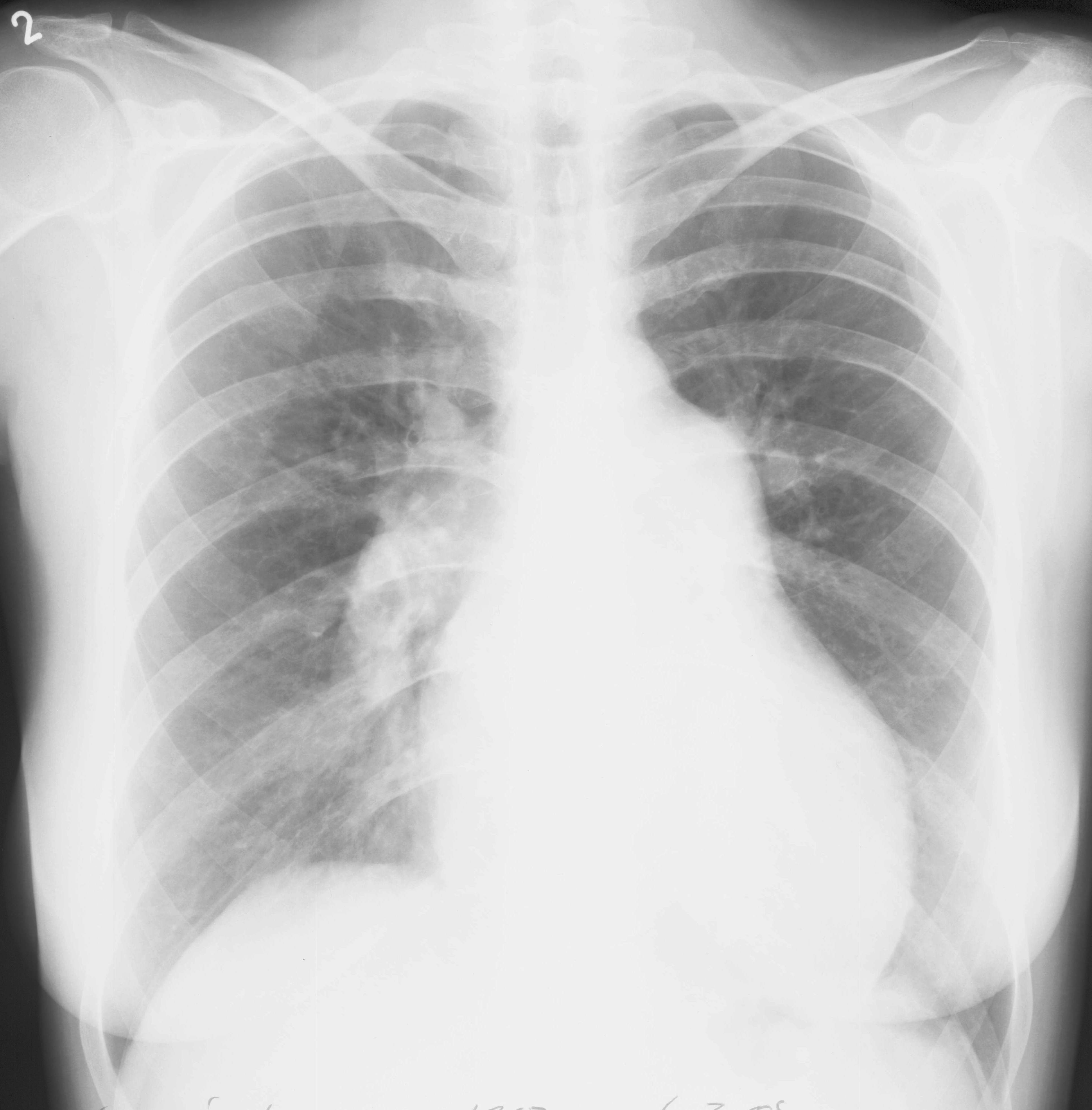
Obr. 5: Skiagram hrudníku u nemocné s pokročilou plicní arteriální hypertenzí. Aortální knoflík je nevýrazný. Levá kontura srdeční je tvořena zvětšenou pravou komorou. Šířka pulmonálního segmentu: 55 mm, šířka sestupné větve pravé plicnice: 23 mm. Laskavě zapůjčil Doc. MUDr. J. Ort, CSc., Radiodiagnostická klinika VFN a 1. LF UK
K určení pokročilosti onemocnění se využívá jednoduchý zátěžový test šestiminutovou chůzí a také vyšetření některých biomarkerů: kyselina močová, troponin, natriuretické peptidy. Výsledky těchto vyšetření umožňují spolu s dalšími klinickými, echokardiografickými a hemodynamickými daty stratifikovat nemocné a tak individualizovat léčbu.
K základním parametrům, které se váží s prognózou onemocnění jak v době stanovení diagnózy tak během sledování při léčbě patří funkční třída podle NYHA, hladina natriuretických peptidů, srdeční index a saturace smíšené žilní krve kyslíkem.
U nemocných s vyšším rizikem PAH je indikován skríning s cílem zachytit onemocnění v málo pokročilém stádiu u oligosymptomatických nebo dokonce asymptomatických nemocných. Echokardiografický skríning PAH je indikován u nemocných se systémovou sklerodermií každý rok, zejména u pacientů s délkou trvání základního onemocnění nad 3 roky, nevýznamnou redukcí plicních objemů, s hodnotou difúzní kapacity pro CO ≤ 60 % a s vyšší hladinou natriuretických peptidů. U ostatních systémových onemocnění pojiva a HIV infekce je skríning PAH indikován v případě manifestace symptomů podezřelých z plicní hypertenze. U prvostupňových příbuzných nemocných s PAH má být ECHO vyšetření prováděno v intervalu 3-5 let, u nemocných s jaterním onemocněním vždy před plánovanou transplantací jater. V případě, že je na základě echokardiografického vyšetření přítomnost plicní hypertenze možná nebo pravděpodobná, je nutno nález ověřit při pravostranné srdeční katetrizaci.
Stanovení definitivní diagnózy PAH patří do rukou expertních center. Nemocní by měli být na tato pracoviště v ideálním případě referováni s podrobným echokardiografickým nálezem, s výsledkem celotělové pletysmografie a difúzní kapacity plic pro CO a s výsledkem ventilační a perfúzní scintigrafie plic.
Léčba
Vedle režimových opatření lze terapeutické možnosti PAH rozdělit na léčbu podpůrnou (antikoagulace, léčba srdečního selhání, oxygenoterapie), léčbu specifickou (vazodilatační léčba blokátory kalciových kanálů, prostanoidy, antagonisté endothelinových receptorů a inhibitory fosfodiesterázy 5) a léčbu nefarmakologickou (balónková atriální septostomie, transplantace plic).
Cílem léčby PAH je nejen zlepšit symptomy, ale také zlepšit prognózu a kvalitu života. Toho lze dosáhnout v případě stabilizace stavu pacienta ve funkční třídě NYHA I nebo II, s normální velikostí a funkcí pravé komory, s hemodynamickými parametry, které svědčí pro normální funkci pravé komory (tlak v pravé síni < 8 mmHg, srdeční index > 2.5-3 l/min/m2), se vzdáleností při testu šestiminutovou chůzí > 380-440 m a s normální hladinou natriuretických peptidů.
Fyzická zátěž je u nemocných s PAH vhodná dle individuální tolerance. Optimální je lehké aerobní cvičení (chůze). Není vhodný pobyt ve vyšších nadmořských výškách (2000 m). Při cestě letadlem je nezbytná inhalace kyslíku. Doporučováno je očkování proti chřipce a proti infekci pneumokokem. Těhotenství je u PAH kontraindikováno. Nutná je účinná antikoncepce. Z hlediska rizika tromboembolismu je akceptovatelná hormonální antikoncepce při současné antikoagulační léčbě. Chirurgické výkony v celkové anestezii trvající déle jak 3 hodiny jsou rizikové.
Diuretika zlepšují symptomy v souvislosti s městnáním při srdečním selhání. Dlouhodobá domácí oxygenoterapie má stejná indikační kritéria jako u plicních onemocnění, délka má být alespoň 15 hodin denně, indikace u nemocných s Eisenmengerovým syndromem je otazná a obecně se nedoporučuje. Digitalis není v současné době běžnou součástí terapie PAH. Chronická antikogulační léčba warfarinem je indikována především u idiopatické, hereditární PAH a u PAH asociované s abúzem anorektik, dále při intravenózní léčbě prostanoidy s cílem snížit riziko trombózy katétru. Cílové INR se má pohybovat kolem 2.
Léčba vysokými dávkami blokátorů kalciových kanálů (nifedipin, diltiazem, amlodipin) je indikována pouze v případě zachovalé vazoreaktivity. Pozitivní vasodilatační test pozorujeme u necelých 11 % nemocných s idiopatickou PAH a podstatně méně často u PAH asociované s ostatními stavy. Dlouhodobé odpovědi na vasodilatační léčbu blokátory kalciových kanálů, která je charakteristická mj. zlepšením symptomů do stádia NYHA I a II, dosáhneme pouze asi u poloviny akutních respondérů (11). Při selhání léčby blokátory kalciových kanálů je nezbytná farmakoterapie prostanoidy, antagonisty receptorů pro endothelin nebo inhibitory fosfodiesterázy-5.
Léčba blokátory kalciových kanálů musí být obezřetně titrována. Léčba retardovaným nifedipinem se zahajuje dávkou 30 mg 2x denně s titrací do dávky 120-240 mg. Léčba diltiazemem začíná na dávce 60 mg 3x denně s cílem 240-720 mg denně. Amlodipin dávkujeme od 2,5 mg denně do cílové dávky více jak 20 mg denně. Účinek léčby musí být pečlivě monitorován včetně hemodynamického vyšetření, a to již 3-4 měsíce od jejího zahájení. Léčba blokátory kalciových kanálů nesmí být náhle přerušena pro riziko rebound fenoménu.
Prostaglandin I2 (prostacyklin) je hlavní produkt metabolismu kyseliny arachidonové v cévním endothelu. Je potentní vazodilatátor v plicní i systémové cirkulaci, dále se vyznačuje vlastnostmi protidestičkovými, antiproliferativními a pozitivně inotropními. U nemocných s PAH je syntéza prostacyklinu v plicních cévách významně snížena.
Syntetický analog prostacyklinu epoprostenol byl poprvé použit pro léčbu PAH v 80. letech 20. století a zůstává jediným lékem užívaným u PAH, který prokazatelně zlepšuje prognózu již během několika měsíců léčby, zejména u pacientů v klinickém stádiu NYHA IV. Limitem léčby je složitý způsob aplikace kontinuálně do centrální žíly vzhledem k velmi krátkému biologickému poločasu preparátu.
Kontinuální nitrožilní infúzí lze podávat také iloprost a treprostinil. Obě tato analoga prostacyklinu mají ve srovnání s epoprostenolem větší stabilitu a delší biologický poločas. Postrádáme však relevantní srovnání zejména jejich dlouhodobého účinku s epoprostenolem.
Treprostinil umožňuje díky své stabilitě subkutánní podání. Dlouhodobý efekt podkožně podávaného treprostinilu je srovnatelný s léčbou intravenózním prostanoidem. Limitem však je lokální bolestivá reakce v místě podkožní infúze. Vyskytuje se až u 85 % léčených a v řadě případů vede k přerušení léčby.
Inhalační léčba u PAH je možná iloprostem a treprostinilem. Hemodynamický účinek obou přípravků je však po inhalaci relativně krátký (30-45 minut u iloprostu a 60-120 minut u treprostinilu). To vede k nutnosti řady inhalací za den (6-9 u iloprostu, 4 u treprostinilu). Přesto zůstává část dne dostatečně léčbou nepokryta. To může vysvětlit otazný dlouhodobý účinek inhalačně aplikovaných prostanoidů.
K perorální léčbě je určen beraprost. Jeho účinek spočívající ve zlepšení vzdálenosti při testu šestiminutovou chůzí je doložen po 3 a 6 měsících léčby, při déletrvající monoterapii však již není přesvědčivý. Beraprost je pro léčbu PAH zaregistrován v Japonsku a Korei, kde je v současné době dostupný také beraprost s prodlouženým uvolňováním.
U PAH je v současné době testován agonista prostacyklinového receptoru. Aktivní metabolit látky NS-304 (selexipag) má biologický poločas zhruba 10 hodin a předpokládá se, že její podání bude spojeno s nižším výskytem nežádoucích účinků typických pro prostanoidy (flush, bolesti hlavy, nauzea, průjmy, bolesti čelistí).
Endothelin-1 (ET-1) hraje zásadní roli v regulaci kardiovaskulárního systému, respiračního systému a v regulaci metabolismu vody a iontů. ET-1 vedle vazokonstrikce indukuje hypertrofii a hyperplázii různých buněk, proliferaci fibroblastů, produkci extracelulární matrix, a rovněž aktivuje mechanismy zánětlivé reakce. Aktivovaný endothelinový systém u PAH lze ovlivnit duální nebo selektivní blokádou endothelinových receptorů.
Existuje rozsáhlá klinická evidence o účinnosti jak duální blokády bosentanem, ale i seletivní blokády receptoru ETA ambrisentanem. Oba preparáty byly testovány v relativně krátkých randomizovaných klinických studiích (maximální doba trvání studie byla 18 týdnů) a byly registrovány pro léčbu PAH. Léčba těmito přípravky vede ke srovnatelnému zlepšení hemodynamických parametrů a ke srovnatelnému prodloužení vzdálenosti při testu šestiminutovou chůzí. Rovněž jednoleté přežití při léčbě antagonisty receptorů pro endothelin je podobné a pohybuje se kolem 95 %.
Jednotliví antagonisté receptorů pro endothelin se liší v bezpečnostním profilu a v interakci se současně podávanými dalšími přípravky. Hepatopatie se vyskytuje častěji při léčbě bosentanem, méně často při léčbě ambrisentanem. Oba léky jsou teratogenní. K retenci tekutin dochází při léčbě bosentanem i ambrisentanem. Bosentan potencuje metabolismus warfarinu a sildenafilu. Lékové interakce mezi ambrisentanem, sildenafilem a warfarinem se nevyskytují.
Macitentan je nový tkáňově specifický duální antagonista receptorů pro endotelin. Byl testován v první dlouhodobé morbi-mortalitní studii u PAH, která při originálním uspořádání na dosud největším souboru pacientů zařazených do klinické studie s PAH (celkem 742) během zhruba 100 týdnů trvání dokumentovala nejen příznivé ovlivnění symptomů, ale také kvality života (zlepšení v 7 z celkem 8 domén dotazníku SF 36) a především jejich prognózy (v dávce 10 mg denně macitentan redukoval riziko zhoršení PAH nebo úmrtí o 45 %, v dávce 3 mg denně byla redukce rizika zhoršení PAH nebo úmrtí o 30 %). Ve srovnání s placebem nevedl macitentan k výraznější indukci hepatopatie nebo k retenci tekutin, častější byl výskyt anemie.
Inhibice degradace cGMP (cyklický guanosin monofosfát) jako druhého posla v regulační kaskádě NO (oxid dusnatý) zesiluje relaxaci hladkých svalových vláken a vazodilataci navozenou cGMP.
Sildenafil je potentní inhibitor fosfodiesterázy 5 (PDE-5) specifické k cGMP. U pacientů s PAH v klinickém stádiu NYHA II a III byl testován v rozsáhlé multicentrické randomizované a placebem kontrolované studii. V léčené skupině při dávkování 3 x denně 20, 40 nebo 80 mg zlepšil po 12 týdnech funkční zdatnost a hemodynamické parametry. Zlepšení funkční zdatnosti přetrvává i po 12 měsících. Ve většině případů však je nutné zvýšit dávku na 3 x denně 50-80 mg. Z dlouhodobého sledování je také zřetelný příznivý vliv sildenafilu na přežívání nemocných, nejedná se však o efekt zaregistrované dávky 20 mg, ale u většiny pacientů o efekt dávky vyšší.
Tadalafil je zaregistrován pro léčbu PAH v dávce 40 mg 1 x denně.
Přímé srovnání účinku sildenafilu a tadalafilu, kromě srovnání akutního hemodynamického efektu, není k dispozici. Sildenafil, na rozdíl od tadalafilu, inhibuje i PDE-1, která se podílí na proliferaci buněk hladkého svalstva v cévní stěně. Není známo, zda tato skutečnost může být klinicky relevantní.
Solubilní guanylát cykláza hraje klíčovou roli v aktivaci cGMP v signální cestě NO. Stimulátory zesilují účinek NO na guanylát cyklázu, zatímco aktivátory mohou indukovat vazodilataci i bez působení NO. V experimentu stimulátory i aktivátory příznivě ovlivňují remodelaci. Riociguat je perorální stimulátor solubilní guanylát cyklázy. Jeho podání vede u PAH k poklesu tlaku v plicnici, ke zvýšení srdečního výdeje, zlepšuje vzdálenost při testu šestiminutovou chůzí, funkční třídu a prodlužuje dobu do klinického zhoršení. Riociguat se v chronickém podání titruje do dávky 2.5 mg 3x denně. Má silné vazodilatační účinky, současné podání s inhibitory PDE-5 je kontraindikováno.
Atriální septostomie je intervenční metoda spočívající ve vytvoření umělé komunikace na úrovni síní se vznikem pravo-levého zkratu. Cílem intervence je zvýšení srdečního výdeje za cenu systémové desaturace. V zemích s dostupnou farmakoterapií je atriální septostomie indikována jako paliativní metoda, případně jako most k transplantaci u nemocných s refrakterním pravostranným srdečním selháním a synkopami při maximální specifické farmakoterapii. V zemích, kde specifická léčba PAH není k disposici, je atriální septostomie často jedinou možnou terapeutickou intervencí. Saturace by neměla po výkonu klesnout o více jak 10 % vstupních hodnot. Vytvoření septostomie o průměru kolem 8 mm zvýší většinou dostatečně srdeční výdej o 20-25 %. Výkon by neměl být indikován při tlaku v pravé síni nad 20 mmHg a při saturaci pod 80 %.
Transplantace plic představuje účinnou léčbu u nemocných v terminálním stádiu PAH po vyčerpání všech ostatních dostupných léčebných možností. Většina center indikuje bilaterální sekvenční transplantaci plic. Transplantace srdce a plic je indikován téměř výlučně u komplexních vrozených srdečních vad. V případě jednoduchých srdečních vad s plicní arteriální hypertenzí se indikuje korekce vady a transplantace plic v jedné době.
Nemocní ve funkčním stádiu NYHA IV mají být zařazeni na čekací listinu k transplantaci plic ihned po stanovení diagnózy a mohou být vyřazeni při zlepšení do funkčního stádia NYHA II. Nemocní ve funkčním stádiu NYHA III jsou indikováni k transplantaci, pokud ani kombinační léčba PAH nevede k výraznějšímu zlepšení. Agresivnější přístup k indikaci transplantace je její zvážení již v případě nedostatečného terapeutického efektu monoterapie.
Za hemodynamická kritéria pro indikaci k transplantaci plic se považuje tlak v pravé síni nad 15 mmHg a srdeční index pod 2 l/min/m2.
Jednoroční přežití po transplantaci plic pro PAH se pohybuje mezi 66 % a 75 %, pětileté přežití kolem 45-50%.
Strategie léčby
Vazodilatační léčba blokátory kalciových kanálů je indikována u pacientů s pozitivním testem akutní plicní vazodilatace, pokud při této terapii zůstávají nemocní dlouhodobě ve funkčním stádiu NYHA I nebo II (Obr.6) . V ostatních případech je indikována léčba přípravky, jejichž účinky jsou nejen vazodilatační, ale také antiremodelační. U nemocných ve funkční třídě NYHA II jsou perspektivně základem léčby vedle klasických antagonistů receptorů pro endothelin ambrisentanu a bosentanu nebo inhibitorů fosfodiesterázy 5 sildenafilu a tadalafilu také macitentan (tkáňově specifický duální antagonista receptorů pro endothelin) a riociguat (stimulátor solubilní guanylátcyklázy). Obdobně je tomu u pacientů ve funkční třídě NYHA III, alternativu představují parenterální prostanoidy. Ve funkčním stádiu NYHA IV je lékem volby intravenózní epoprostenol, případně další analoga prostacyklinu, antagonisté receptorů pro endothelin a inhibitory fosfodiesterázy 5. V případě nedostatečné terapeutické odpovědi je indikována kombinační léčba, případně balónková atriální septostomie a transplantace plic.
Kritéria nedostatečné odpovědi na léčbu po 3-4 měsících od jejího zahájení představují přetrvávající funkční třída podle NYHA III případně IV, výrazně snížená zátěžová kapacita, nízký srdeční výdej, vysoký tlak v pravé síni, vysoká hladina natriuretických peptidů, echokardiografické známky dysfunkce pravé komory a potřeba eskalace léčby.
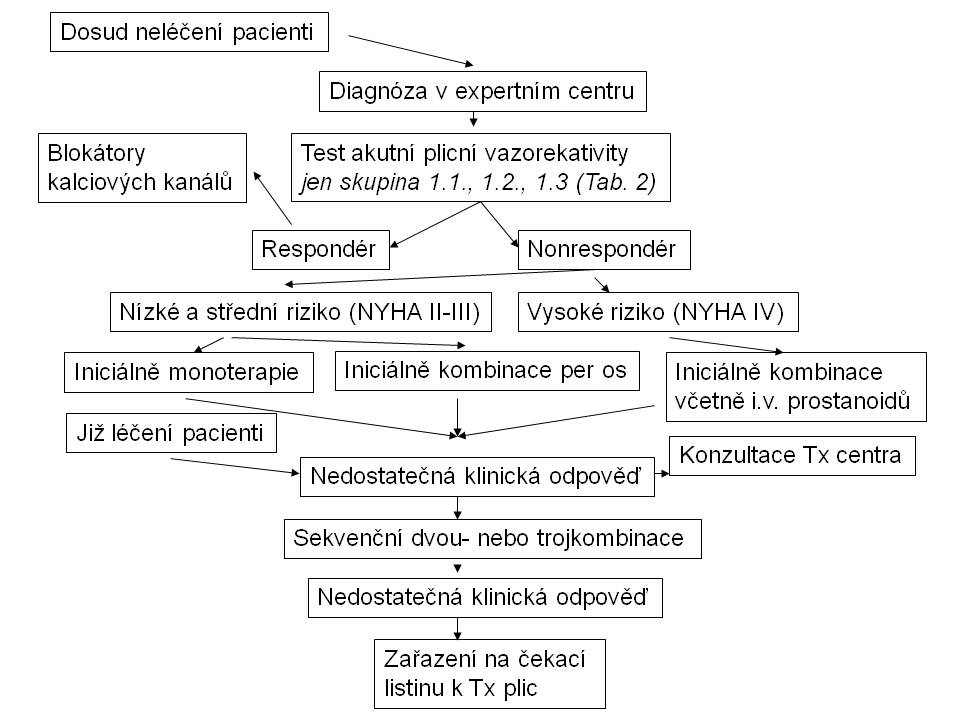
Obr. 6: Terapeutický algoritmus plicní arteriální hypertenze
Plicní venookluzivní nemoc a plicní kapilární hemangiomatóza (skupina 1´)
Plicní venookluzivní nemoc a plicní kapilární hemangiomatóza jsou vyčleněny ze skupiny PAH pro svoji odlišnou patofyziologii a léčbu. V histologickém obrazu jsou přítomny typické hypertenzní změny na cévách, ale zároveň i nálezy charakteristické pro postkapilární plicní hypertenzi. Klinický obraz je od PAH obtížně odlišitelný. Výskyt je raritní.
Diagnostika
Pro pacienty s plicní venookluzivní nemocí je charakteristický výskyt hlubší hypoxemie a významnější redukce difúzní kapacity pro CO, než bývá u pacientů s PAH. Klíčovou vyšetřovací metodou je HRCT, kde bývá nálezem subpleurálně ztluštění interlobulárních sept, denzity typu mléčného skla centrilobulárně a mediastinální lymfadenopatie.
Léčba
Vazodilatační léčba, a zejména léčba prostanoidy, je u těchto nemocných zatížena vysokým rizikem rozvoje plicního edému. Indikace atriální septostomie je většinou omezena hypoxemií. Léčbou volby je transplantace plic, k níž mají být nemocní indikováni ihned po stanovení dia¬gnózy.
Perzistující plicní hypertenze novorozenců-PPHN (skupina 1´´)
Perzistující plicní hypertenze novorozenců (PPHN) byla dříve řazena do skupiny PAH. Vzniká jako důsledek poruchy relaxace plicních cév po narození a vede ke zkratovému proudění neoxygenované krve z plicní do systémové cirkulace přes otevřenou tepennou dučej nebo foramen ovale, nejčastěji u donošených nebo přenošených novorozenců. Vyšší riziko je u matek, které zejména po 20. týdnu gravidity užívaly inhibitory zpětného vychytávání serotoninu. Příčinou rozvoje PPHN mohou být poruchy vývoje (diafragmatická hernie, plicní hypoplázie, renální ageneze, cévní malformace) nebo poruchy adaptace (nejčastěji při aspiraci mekonia). Vyskytuje se s frekvencí 1-2 případy na 1000 novorozenců. Mortalita je vysoká (10-20 %).
Diagnostika
Pro PPHN je charakteristická hypoxémie špatně reagující na oxygenoterapii. Bývá vyšší hladina natriuretických peptidů. Nezbytné je vyloučit vrozenou cyanotickou srdeční vadu.
Léčba
K základním terapeutickým nástrojům u PPHN patří oxygenoterpie, umělá plicní ventilace, podání NO a mimotělní podpora pomocí extrakorporální membránové oxygenace. Zkouší se rovněž vazodilatační léčba podobně jako u PAH.
Plicní hypertenze při postižení levého srdce (skupina 2)
Pro definici plicní hypertenze u onemocnění levého srdce je rozhodující diastolický tlakový gradient (rozdíl mezi diastolickým tlakem v plicnici a tlakem v zaklíněné plicnici). Normální hodnota je 1-2 mmHg, abnormální hodnoty jsou > 5 mmHg, hodnoty ≥ 7 mmHg jsou prognosticky závažné a hodnoty ≥ 10 mmHg svědčí pro významný podíl prekapilární složky.
Plicní hypertenzi s tlakem v zaklíněné plicnici > 15 mmHg a diastolickým gradientem < 7 mmHg lze označit jako izolovanou postkapilární. Plicní hypertenzi s tlakem v zaklíněné plicnici > 15 mmHg a diastolickým gradientem ≥ 7 mmHg lze označit jako kombinovanou postkapilární a prekapilární.
Plicní hypertenze nezřídka provází onemocnění myokardu nebo chlopní levého srdce (postkapilární plicní hypertenze, plicní žilní hypertenze). Podíl tohoto typu plicní hypertenze mezi pacienty s nevysvětlenou plicní hypertenzí je vysoký a stoupá s věkem. Celkově se v populaci jedná o nejčastější chronickou plicní hypertenzi. U pacientů se systolickou dysfunkcí levé komory srdeční a také u pacientů s diastolickou dysfunkcí levé komory srdeční bývá plicní hypertenze přítomna až u 70 % nemocných. U kandidátů transplantace srdce má určitý stupeň plicní hypertenze většina nemocných. Plicní hypertenze u srdečních onemocnění je zprvu vždy typicky postkapilární. Tlak v zaklínění a tlak v plicnici stoupá v počátečních stádiích onemocnění lineárně. Později však dochází u některých nemocných k nelineárnímu nárůstu tlaku v plicnici, především díky vazokonstrikci a remodelaci plicních cév, zvyšuje se také transpulmonální a diastolický tlakový gradient. Naopak tlak v zaklínění u nemocných s diastolickým srdečním selháním léčených diuretiky nemusí být v klidu zvýšen.
U plicní hypertenze při onemocnění levého srdce bývá zvětšený objem levé síně (> 40 ml/m2). K rizikovým faktorům zejména diastolického srdečního selhání patří věk nad 65 let, přítomnost arteriální hypertenze, fibrilace síní, diabetu, obezity, anamnéza přechodné plicní kongesce na snímku hrudníku a symptomatická odpověď na diuretickou léčbu.
Přítomnost plicní hypertenze u levostranného srdečního selhání značně nepříznivě ovlivňuje prognózu. Mortalita těchto pacientů je až 3x vyšší než u nemocných bez plicní hypertenze.
Léčba
U nemocných s plicní žilní hypertenzí je základem léčby dobrá kontrola systémové arteriální hypertenze, dále adekvátní léčba základního onemocnění levého srdce (diuretika, inhibitory angiotenzin konvertujícího enzymu, beta blokátory, inhibitory fosfodiesterázy, digitalis, korekce významné chlopenní vady). I při normalizaci plnících tlaků levé komory však u řady nemocných plicní hypertenze přetrvává. Jde zejména o pacienty s plicní hypertenzí, jejíž závažnost neodpovídá základnímu onemocnění (tlak v zaklínění do 22 mmHg, střední tlak v plicnici nad 35-40 mmHg, transpulmonální gradient nad 18-20 mmHg). Jako logická se pak nabízí otázka specifické léčby podobně jako u PAH.
Specifická vazodilatační léčba antagonisty receptorů pro endothelin a prostanoidy u nemocných s plicní žilní hypertenzí byla dosud zkoušena neúspěšně. Příčinou je zřejmě zlepšení žilního návratu a zvýšení srdečního výdeje při terapii. To může vést k dalšímu nárůstu levostranných plnících tlaků a k selhání levé komory.
V poslední době však existují sdělení o příznivém účinku sildenafilu na redukci plicní cévní rezistence, zejména u kandidátů transplantace srdce. Přetrvávající vyšší plicní cévní rezistence na vrcholu vazodilatačního testu je totiž zásadní překážkou transplantace srdce.
U nemocných rezistentních k farmakoterapii a nevhodných k resynchronizační terapii lze indikovat mechanické srdeční podpory, které slouží ke zvládnutí akutního zhoršení nebo jako most k transplantaci. Zlepšení symptomů je u těchto nemocných provázeno poklesem tlaku v plicnici.
Pokles plicní cévní rezistence po transplantaci srdce závisí na její tíži před výkonem.
Plicní hypertenze při plicních onemocněních a/nebo při hypoxémii (skupina 3)
Plicní hypertenze u respiračních onemocnění je druhou nejčastější plicní hypertenzí po plicní hypertenzi při postižení levého srdce. Typicky se jedná o prekapilární plicní hypertenzi, která je u většiny pacientů lehká, ale její přítomnost je závažným negativním prediktorem prognózy.
U chronických plicních onemocnění se za normotenzi v plicnici považují hodnoty středního tlaku v plicnici < 25 mmHg. Za plicní hypertenzi jsou považovány hodnoty středního tlaku v plicnici 25-34. Situace, kdy střední tlak v plicnici dosahuje hodnot ≥ 35 mmHg nebo je ≥ 25 mmHg a současně je srdeční index < 2 l/min/m2, se označuje jako těžká plicní hypertenze.
Těžká plicní hypertenze se u respiračních onemocnění vyskytuje spíše vzácně, maximálně u několika procent nemocných. Např. v populaci pacientů léčných dlouhodobou domácí oxygenoterapií je takových nemocných asi 1 %. Příčinou této těžší plicní hypertenze může být jiná koincidující klinická jednotka charakteristická syndromem plicní hypertenze (např. CTEPH, spánková apnoe), která může být sama o sobě dobře terapeuticky ovlivnitelná. Příčinou mohou být rovněž remodelační změny v oblasti plicních arteriol. Proto by nemocní s respiračním onemocněním a těžkou plicní hypertenzí měli být rovněž vyšetřeni ve specializovaném centru.
Diagnostika
K základním vyšetřovacím metodám patří echokardiogrefie, snímek hrudníku CT (HRCT) plic a vyšetření plicních funkcí. Ventilační a perfúzní scintigrafie plic je nezbytná k vyloučení CTEPH. Pravostranná srdeční katetrizace je indikována zejména v případě podezření na těžkou plicní hypertenzi a při úvahách o transplantaci plic.
Pro sekundární plicní hypertenzi při plicním onemocnění svědčí nález ventilační poruchy (FEV1 < 60 % náležitých hodnot, FVC < 70 % náležitých hodnot), hypoxémie a charakteristický nález na HRCT plic. Pro jinou příčinu, zejména těžké plicní hypertenze, svědčí absence významnější ventilační poruchy (FEV1 > 60 % náležitých hodnot, FVC > 70 % náležitých hodnot), nepřítomnost hypoxémie a významnějšího nálezu na HRCT plic.
Léčba
Chronická obstrukční plicní nemoc je v 8-10 % komplikována plicní hypertenzí, většinou lehkou. Terapie předpokládá adekvátní léčbu základního onemocnění. Kyslík je jediné dostupné vazodilatans selektivní pro plicní oběh. Dlouhodobá oxygenoterapie (minimálně 14-16 hodin) brání progresi plicní hypertenze a zejména zlepšuje prognózu. Vzhledem k tomu, že plicní hypertenze je u respiračních onemocnění většinou mírná, hemodynamické změny zřejmě nejsou hlavní příčinou zlepšení prognózy. Oxygenoterapie vede především ke zlepšení oxygenace tkání, snad také příznivě ovlivňuje remodelaci plicních cév. Léčba kyslíkem vede rovněž k poklesu hematokritu a v důsledku zlepšení reologických vlastností krve k poklesu cévní rezistence a ke zlepšení funkce pravé komory. Venepunkce vede ke zlepšení hemodynamiky a ke krátkodobému zlepšení tolerance zátěže. Indikována je u nemocných s hematokritem vyšším než 55, cílem je dosažení hematokritu kolem 50. Účinek specifické vazodilatační terapie není přesvědčivě doložen. Transplantace jedné plíce nebo transplantace obou plic je indikována u nemocných v terminálním stádiu plicních onemocnění. Již transplantace jedné plíce může normalizovat hemodynamické poměry. Jednoleté přežití se pohybuje kolem 60 %, pětileté přežití kolem 40 %.
Někteří pacienti s emphyzémem mohou profitovat z volum reduktivní plicní resekce. Nemocní s významnější plicní hypertenzí však nejsou vhodnými kandidáty. Existují rozporuplné údaje o efektu tohoto výkonu na plicní hemodynamiku. U nemocných s preexistující plicní hypertenzí dochází pooperačně často k nárůstu tlaku v plicnici. Spíše ojedinělé jsou údaje o významnějším zlepšení funkce pravé komory po výkonu.
Intersticiální plicní procesy jsou relativně častou příčinou plicní hypertenze, 60-70 % pacientů má v terminálním stádiu své nemoci známky plicní hypertenze, která je typicky lehká. Její léčba je obtížná, i při adekvátní terapii základního onemocnění perzistuje, nezřídka progreduje a vede k selhání pravého srdce. Vedle dlouhodobé domácí oxygenoterapie přichází v úvahu také transplantace plic. Efekt specifické vazodilatační terapie není prokázán.
Syndrom obstrukční spánkové apnoe (OSA) je charakterizován mnohočetnými apnoickými pauzami, epizodami hlasitého chrápání a excesivní denní spavostí. Plicní hypertenzi má asi 12 % pacientů s OSA. Bývá nejčastěji mírná a léčbu nevyžaduje. Její přítomnost je však dokladem manifestace dalších komplikací OSA (arteriální hypertenze, poruchy srdečního rytmu, ischémie myokardu) a tedy nutnosti jeho léčby (režimová opatření, léčba kontinuálním přetlakem, chirurgická léčba).
U sarkoidózy se plicní hypertenze vyskytuje asi u 5 % nemocných a představuje významný negativní prediktor prognózy. Příčina může být různorodá (fibrotická ablace plicní vaskulatury při intersticiálním postižení, mediastinální fibróza, komprese velkých plicních cév při lymfadenopatii, plicní žilní hypertenze při dysfunkci levého srdce a vaskulopatie při granulomatózním postižení). Léčba zahrnuje terapii základního onemocnění, oxygenoterapii a přáípadně indikaci k transplantaci plic. Evidence pro specifickou vazodilatační léčbu chybí.
Chronická tromboembolická plicní hypertenze (skupina 4)
CTEPH souvisí s akutní plicní embolií. Její vznik však nelze vysvětlit pouze obliterací plicních cév organizovanými nerekanalizovanými tromby, které u většiny pacientů uzavírají více než 40 % plicních cév. Akutní plicní embolie má zřejmě úlohu spouštěče kaskády dějů zahrnující také remodelaci plicních cév a sekundární trombózy in situ, které mohou ústit v rozvoj CTEPH. Je velmi pravděpodobné, že podobně jako u PAH existuje genetická dispozice pro vznik CTEPH, kterou zatím neznáme. Také ostatní molekulární mechanismy podílející se na rozvoji CTEPH jsou pravděpodobně velmi podobné jako u ostatních typů těžké chronické plicní hypertenze. Tyto úvahy otevírají možnost podobného terapeutického přístupu u PAH a některých nemocných s CTEPH. Anamnéza plicní embolie je němá až u 25 % nemocných s CTEPH (31).
Výskyt CTEPH není přesně znám. Jednou z příčin je jistě vztah CTEPH k akutní plicní embolii, jejíž výskyt v populaci rovněž přesně neznáme. Proti tradovaným odhadům, že k rozvoji CTEPH dojde u 0,1–0,5 % nemocných, kteří přežijí epizodu akutní plicní embolie, se ukazuje, že výskyt CTEPH bude pravděpodobně vyšší, nejspíše kolem 2–4 %.
K rizikovým faktorům vzniku CTEPH patří vyšší hladina antifosfolipidových protilátek, přítomnost lupus antikoagulans, opakované příhody plicní embolie, neznámý zdroj plicní embolie, rozsáhlejší perfuzní defekty, anamnéza maligního onemocnění, hypotyreóza, infekce kardiostimulační soustavy, přítomnost ventrikuloatriální spojky pro léčbu hydrocefalu, chronické záněty (osteomyelitida, nespecifické střevní záněty) a splenektomie.
Údaje o prognóze neléčené CTEPH pocházejí ze starších prací, neboť v současné době je většina nemocných referována k chirurgické léčbě.
Čeští autoři publikovali v roce 1982 výsledky chronického sledování 76 nemocných po akutní plicní embolii, kteří neměli jiné kardiovaskulární onemocnění (34). Doba sledování byla 1 až 15 let. Klíčový prediktor prognózy byla výše středního tlaku v plicnici. Zřetelně horší prognózu měli nemocní se středním tlakem v plicnici nad 30 mmHg a zejména ti, kteří měli známky pravostranného srdečního selhání. Ve skupině se středním tlakem v plicnici nad 50 mmHg přežilo 2 roky méně než 20 % pacientů. Naopak prognóza nezávisela na počtu, rozsahu a lokalizaci plicní embolie, věku a léčbě.
Obdobné jsou výsledky sledování 49 nemocných s CTEPH léčených pouze antikoagulační léčbou.
Je mimo jakoukoli pochybnost, že u nemocných s přetrvávající plicní hypertenzí po plicní embolii jejich onemocnění progreduje a bez léčby vede k pravostrannému srdečnímu selhání a úmrtí. V současné době je prognóza nemocných významně zlepšena především díky dostupné chirurgické léčbě.
Diagnostika
U nemocných s podezřením na plicní hypertenzi používáme jako základní vyšetření k její detekci echokardiografii s dopplerovským vyšetřením. Pokud zjišťujeme při akutní plicní embolii systolický tlak v plicnici vyšší než 50 mmHg, velmi pravděpodobně již jde o CTEPH. K odhalení rizika rozvoje CTEPH by měli být echokardiograficky vyšetřeni všichni pacienti do asi 6 týdnů po epizodě akutní plicní embolie a pak zejména ti, u nichž dojde během dalšího období k manifestaci symptomů podezřelých z plicní hypertenze.
V případě přítomnosti ECHO známek plicní hypertenze, pokud není nález vysvětlitelný srdečním nebo plicním onemocněním, indikujeme ventilační a perfuzní scintigrafii plic, která je v této indikaci výrazně výtěžnější než CT angiografie (obr.7). Normální perfuzní scintigrafie prakticky vylučuje CTEPH (obr. 8).
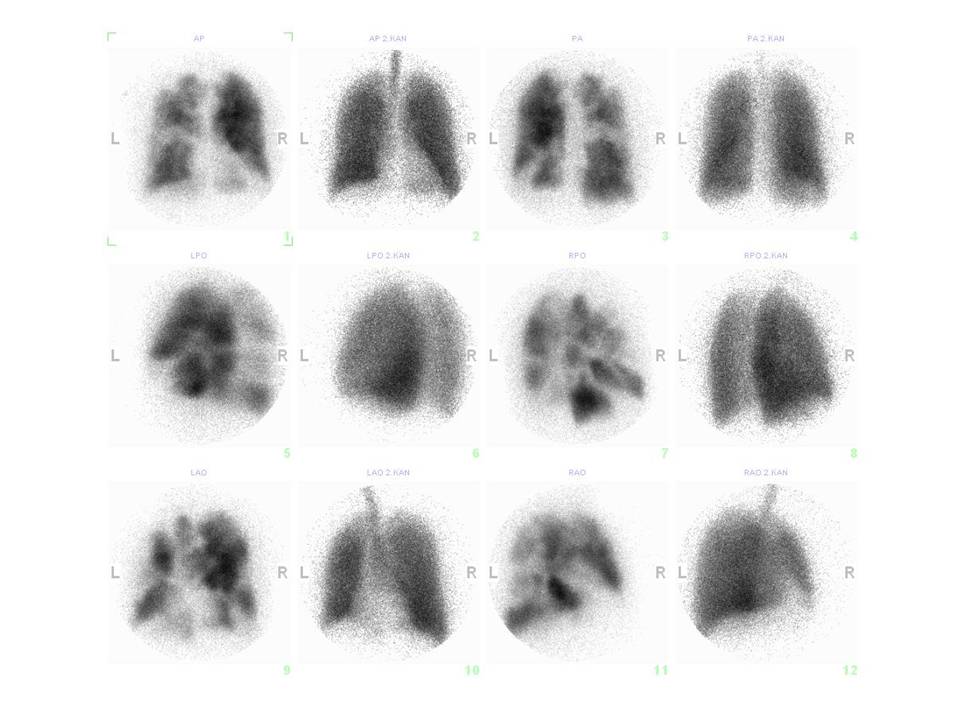
Obr. 7: Četné oboustranné segmentární a subsegmentární defekty na perfúzním scintigramu plic a normální ventilační scintigram plic u nemocného s chronickou tromboembolickou plicní hypertenzí.
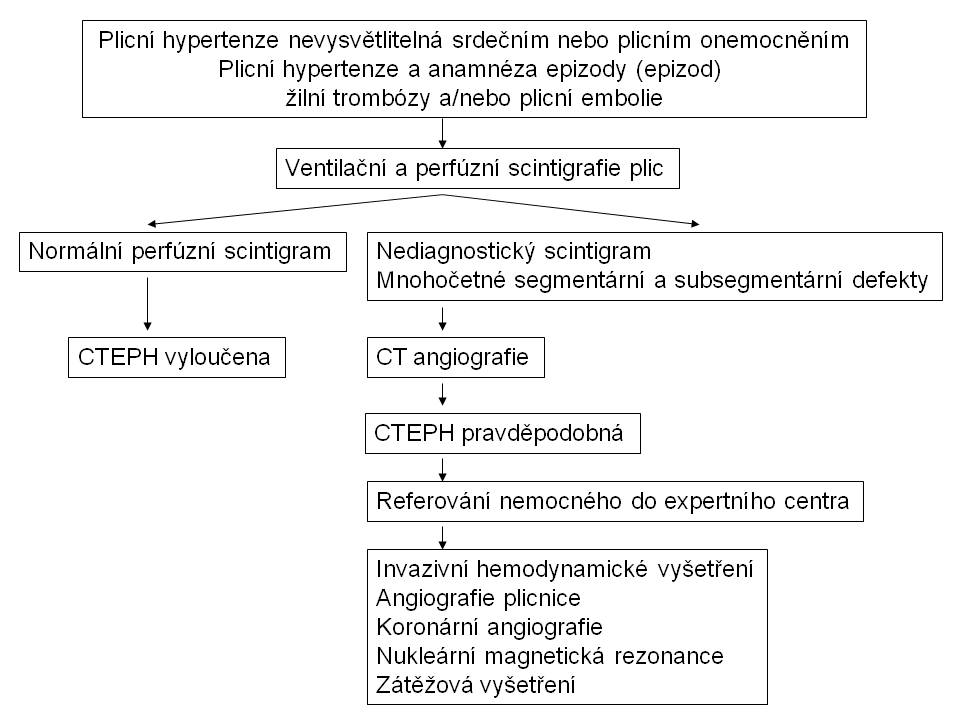
Obr. 8: Diagnostický algoritmus u chronické tromboembolické plicní hypertenze
Laskavě zapůjčil Prim. MUDr. Jozef Kubinyi, Ph.D., Ústav nukleární medicíny VFN a 1.LF UK, Praha
Pokud je scintigrafické vyšetření nediagnostické nebo prokazuje vícečetné oboustranné segmentární, event. subsegmentární defekty, je indikována CT angiografie. V případě nálezu vysoce podezřelého z CTEPH je nezbytné kontaktovat centrum, které se zabývá léčbou tohoto onemocnění, a diskutovat další diagnostický postup. Invazivní vyšetření, které má být provedeno ve specializovaném centru, zahrnuje pak vedle angiografie plicnice rovněž hemodynamické vyšetření a koronární angiografii.
Konvenční angiografie plicnice zůstává nadále zásadním vyšetřením k definitivnímu stanovení diagnózy CTEPH a k rozhodnutí o způsobu léčby (obr. 9). Provádí se současně s pravostrannou srdeční katetrizací. Nevýhodou vyšetření je radiační zátěž, nutnost podání kontrastní látky s možnými alergickými komplikacemi, omezená dostupnost a riziko úmrtí, které stoupá se závažností plicní hypertenze. Angiografie plicnice u CTEPH zahrnuje sérii projekcí v jednotlivých fázích perfúze od vstřiku kontrastní látky do plicnice až do žilní fáze včetně parenchymografie sloužící k zobrazení neperfundovaných oblastí.
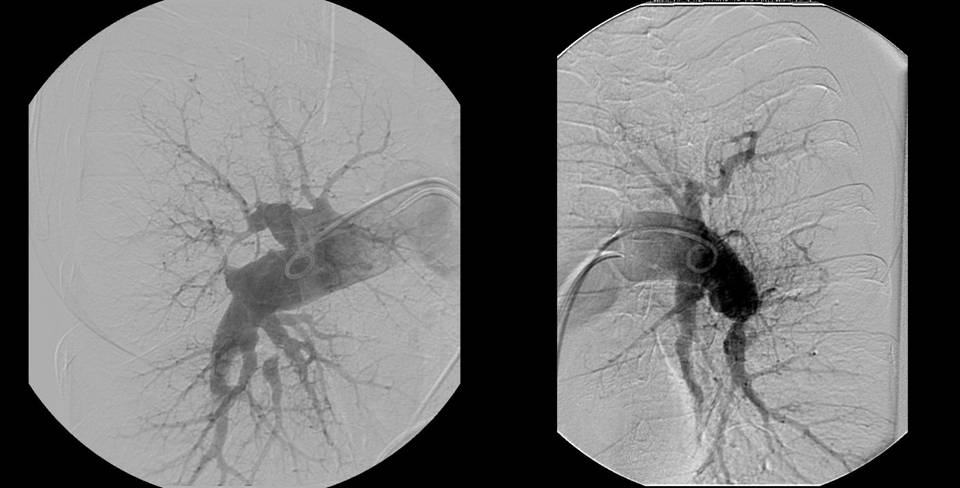
Obr. 9: Typický angiogram u nemocného s chronickou tromboembolickou plicní hypertenzí s oboustranným centrálním postižením.
Laskavě zapůjčil MUDr. Samuel Heller, Ph.D., II. interní klinika kardiologie a angiologie VFN a 1.LF UK, Praha
U CTEPH se popisuje se 5 charakteristických angiografických nálezů, které se různě kombinují:
- stop kontrastní látky způsobený obstrukcí větve plicnice. Tento nález, pokud je přítomen proximálně ve kmeni plicnice, může mimikovat její agenezi
- příčné pruhy uvnitř cévního lumen, které připomínají struny
- nepravidelnosti cévní stěny
- stenózy s poststenotickými dilatacemi
- absence segmentárních nebo lobárních větví plicnice s odpovídajícími defekty v parenchymu
Angiografii kompletizujeme selektivní koronární angiografií. Koincidující ischemická choroba není u nemocných s CTEPH vzácností a její léčbu je nutno zahrnout do terapeutických úvah CTEPH.
Pravostranná srdeční katetrizace s hemodynamickým vyšetřením je nedílnou součástí invazivního vyšetření CTEPH. Vyšetření umožňuje objektivně určit tlak v pravé síni, tlaky v pravé komoře, plicnici a v zaklínění, změřit minutový srdeční výdej a dopočítat plicní cévní rezistenci. Existují práce, které se pokoušejí na základě analýzy křivky v zaklínění plicních kapilár určit podíl centrální trombotické obstrukce plicních cév odstranitelné chirurgicky a periferní remodelace plicních arteriol, která je chirurgicky neovlivnitelná. V tomto základním bodě totiž většina ostatních vyšetřovacích metod selhává.
Nukleární magnetická rezonance umožňuje posuzovat nejen morfologické, ale rovněž funkční parametry plicního oběhu. Vyšetření se považuje za metodu volby pro určení rozměrů pravostranných srdečních oddílů a hmotnosti myokardu, navíc se jedná o metodu neinvazivní bez radiační záteže, jejíž dostupnost se postupně zvyšuje. Nálezy při angiografii pomocí magnetické rezonance do segmentární úrovně dobře korelují s CT angiografií. Periferněji je nadřazena CT angiografie a konvenční angiografie plicnice.
Léčba
Po stanovení diagnózy CTEPH je indikována dlouhodobá antikoagulační léčba s cílovým INR 2,5 až 3 (obr. 10). Někdy při léčbě dochází ke zlepšení hemodynamiky a funkční zdatnosti. Pokud po tříměsíční antikoagulační léčbě přetrvává významnější plicní hypertenze, je nezbytné definitivní vyšetření s otázkou vhodné léčebné strategie.
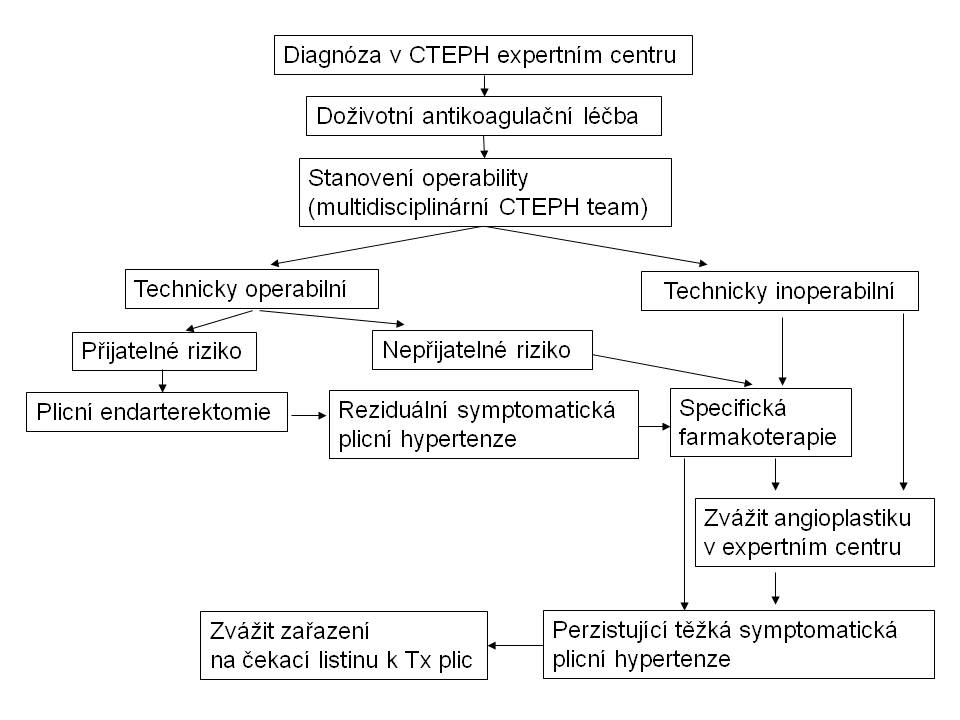
Obr. 10: Terapuetický algoritmus u chronické tromboembolické plicní hypertenze
Při úvahách o kauzální léčbě CTEPH je zcela zásadní průkaz lokalizace obstrukce plicního cévního řečiště. Léčebnou metodou volby u CTEPH je endarterektomie plicnice (PEA) (obr. 11). Principem operace není embolektomie, ale endarterektomie, tedy odstranění organizovaného fibrotizovaného trombu s částí cévní stěny plicnice. Výkon se provádí ze sternotomie v mimotělním oběhu a v hluboké hypotermii. Pro úspěch operace je nezbytná vizualizace distálních větví plicnice. Ta je v případě CTEPH komplikována výrazným kolaterálním přítokem z bronchiálních tepen. Proto se vlastní endarterektomie, která trvá 20-30 minut vpravo a 20-30 minut vlevo, provádí v kompletní cirkulační zástavě. Jako ochrana mozku slouží hypotermie. Operativa je celosvětově soustředěna do několika multidisciplinárních center, včetně jednoho centra v České republice (Kardiocentrum Všeobecné fakultní nemocnice v Praze). Indikováni jsou symptomatičtí nemocní (zejména ve stadiu NYHA III a IV) s chirurgicky dosažitelnou trombotickou obstrukcí plicních cév a plicní cévní rezistencí nad 4 WU. Po výkonu je nutná doživotní antikoagulace. Akceptovatelná mortalita PEA je do 10 %. Riziko výkonu zvyšuje splenektomie, ventrikulo-atriální spojky pro léčbu hydrocefalu, plicní cévní rezistence nad 12 WU. Po operaci je nutno sledovat nemocného alespoň 12 měsíců v operujícím centru, neboť maximální efekt PEA lze očekávat zhruba do 6 měsíců od výkonu. U většiny nemocných dochází k významnému poklesu tlaku v plicnici, často k jeho normalizaci, k vzestupu srdečního výdeje, ke zlepšení výkonnosti, symptomů a dlouhodobé prognózy (VID9+stacion9, VID10+stacion10). Reziduální plicní hypertenze po operaci má arbitrálně stanovenou hemodynamickou definici více jak 3.75 WU.
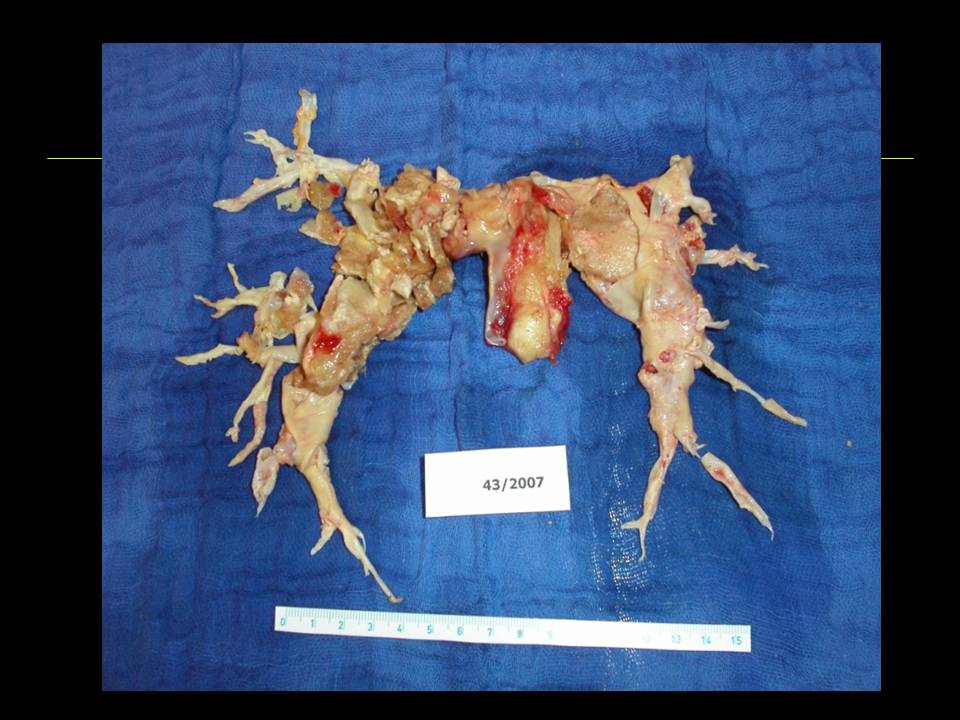
Obr. 11: Endarterium odstraněné u nemocného s těžkou chronickou tromboembolickou plicní hypertenzí při endarterektomii plicnice. Výkon vedl k normalizaci tlaků v plicnici. Laskavě zapůjčil Prof. MUDr. J. Lindner, CSc., II. chirurgická klinika kardiovaskulární chirurgie VFN a 1. LF UK v Praze
U nemocných s reziduální plicní hypertenzí, stejně jako u pacientů inoperabilních pro periferní postižení může být perspektivně indikována farmakoterapie riociguatem (stimulátor solubilní guanylátcyklázy). Jedná se o první látku, která v prospektivní randomizované placebem kontrolované studii prokázala signifikantní zlepšení tolerance zátěže a hemodynamiky.
Transplantace plic může představovat řešení pro nemocné po neúspěšné PEA nebo refrakterní k farmakoterapii, popř. nevhodné k PEA. Dlouhodobé přežívání po transplantaci je podstatně horší než po PEA.
Balonková angioplastika není alternativou PEA a je stále spíše metodou experimentální.
Implantace kaválních filtrů má být přísně individualizovaná a je opodstatněná především u pacientů s očekávanou komplikovanou titrací antikoagulační léčby.